AZD 6482/KIN-193
货号:
IA1950
品牌:
Jinpan
暂无详情
产品简介
| MDL | MFCD00135663 |
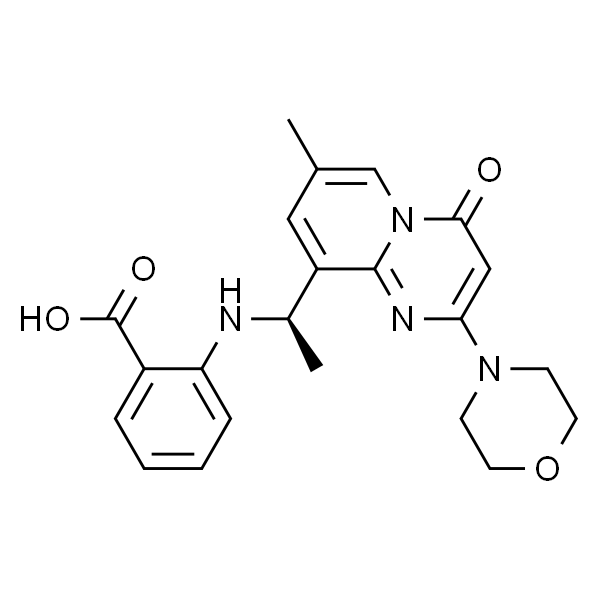
| 别名 | 2-[[(1R)-1-[7-甲基-2-(4-吗啉)-4-氧代-4H-吡啶并[1;2-a]嘧啶-9-基]乙基]氨基]苯甲酸;KIN-193 |
| 英文名称 | AZD 6482/KIN-193 |
| CAS | 1173900-33-8 |
| 分子式 | C22H24N4O4 |
| 分子量 | 408.45 |
| 纯度 | ≥99% |
| 单位 | 瓶 |
| 生物活性 | AZD 6482 是一种有效的选择性 p110β 抑制剂,IC50 为 0.69 nM。[1] |
| In Vitro | 体外激酶试验证明,AZD 6482(KIN-193)在抑制p110β激酶活性方面具有很高的效力(IC50为0.69 nM),分别比p110α,p110δ和p110γ同种型具有200,20和70倍的选择性。 。 AZD 6482还表现出比PI3K-C2β和DNA-PK高约80倍的选择性,比其他磷脂酰肌醇-3激酶相关激酶(PIKKs)高1,000倍。使用KinomeScan方法对一组433种激酶的AZD 6482的抑制剂 – 激酶相互作用分析表明AZD 6482在其与PI3K的相互作用中具有高度选择性。为了确定AZD 6482是否选择性地靶向PTEN缺陷肿瘤,使用高通量肿瘤细胞系谱分析在大量422个癌细胞系上测试AZD 6482对细胞增殖的影响。 35%具有PTEN突变的细胞系(57个中的20个)和16%具有野生型PTEN的细胞系(365个中的58个)对AZD 6482敏感,阈值EC50 <5μM[1]。 |
| In Vivo | 为了确定AZD 6482(KIN-193)在体内肿瘤中的药效学,大鼠成纤维细胞(Rat1)细胞被工程化以表达p53DD,p53的显性失活突变体和组成型激活的myr-p110β(Rat1-CA-p110β) )使这些细胞能够在小鼠中形成异种移植肿瘤。为了比较,还产生了表达p53DD和myr-p110α(Rat1-CA-p110α)的同基因Rat1细胞系。将Rat1-CA-p110α和Rat1-CA-p110β细胞皮下引入无胸腺小鼠的对侧腹侧,使得由活化的p110α或p110β驱动的肿瘤暴露于相同的条件,并且可以消除对动物与动物变异性的关注。 。当肿瘤达到约500mm 3的体积时,携带肿瘤的小鼠接受单次IP注射AZD 6482(10mg/kg)。 AZD 6482的血浆浓度在注射后1小时最高,并且在4小时后降至不可检测的水平。 CA-p110α-和CA-p110β-驱动的肿瘤中的AZD 6482浓度平行于血浆浓度。在AZD 6482注射后不同时间点收集的肿瘤裂解物的分析显示,在Rat1 6482注射Rat1 64-CA1-p110β肿瘤后1小时,AKT的磷酸化显著降低,但在Rat1-CAp110α肿瘤中保持不变[1]。 |
| 激酶实验 | AZD 6482(KIN-193)的分布浓度为10μM,针对433种激酶的不同组。主屏幕命中的分数报告为DMSO对照的百分比(%对照)。对于未显示得分的激酶,未检测到可测量的结合。分数越低,Kd可能越低,因此零分表示强烈的命中。分数与命中概率有关,但严格来说不是亲和力测量。在10μM的筛选浓度下,小于10%的得分意味着假阳性概率小于20%并且Kd最可能小于1μM。 1-10%的分数意味着假阳性概率小于10%,尽管难以从单点主屏幕分配定量亲和力。得分小于1%意味着假阳性概率小于5%,Kd最可能小于1μM[1]。 |
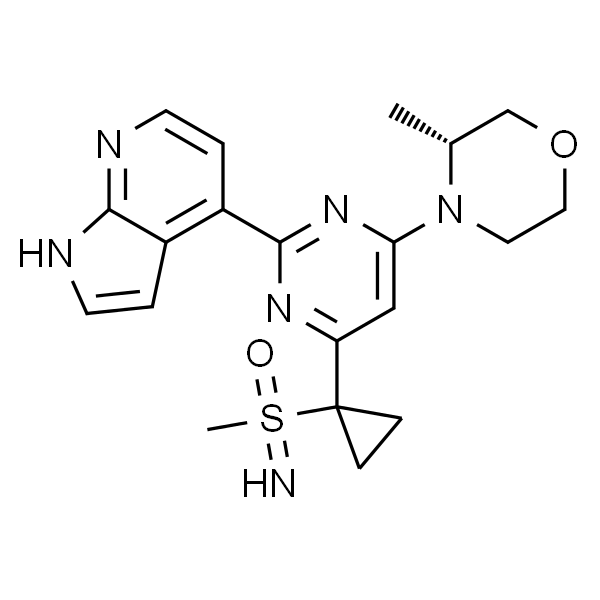
| SMILES | O=C1N2C(C([C@H](NC3=CC=CC=C3C(O)=O)C)=CC(C)=C2)=NC(N4CCOCC4)=C1 |
| 靶点 | PI3K |
| 动物实验 | 小鼠[1]大约6-8周龄雌性裸鼠皮下注射Rat1-Myr-HA-p110α(Rat1-CAp110α)细胞(40%基质胶中1×106个细胞)的一侧(位点1)和Rat1 -Myr-HA-p110β(Rat1-CAp110β)细胞(在10%基质胶中的0.5×106细胞)在对侧胁腹(位点2)。当肿瘤生长至~500mm 3时,通过腹膜内注射用7.5%NMP,40%PEG400,52.5%dH 2 O,0.1mL/10g体重和10mg/kg配制的AZD 6482给予小鼠一次。在化合物给药后0,1,4,8和24小时收集肿瘤,并通过直接心脏穿刺获得血液样品。分离血清并储存在-80℃。通过DMPK组的LC-MS/MS分析评估血清和肿瘤样品中的药物浓度。 |
| 细胞实验 | 确定细胞活力。简而言之,将细胞接种在含有5%FBS的培养基中,密度确保整个药物处理过程中细胞生长(对于大多数细胞系约为15%)。接种后24小时开始药物处理并持续72小时。将细胞固定并使用红色荧光DNA染色剂Syto60染色。通过取减去背景(无细胞孔)后来自药物处理的孔的相对荧光强度与未处理的孔的比率来计算相对细胞数。九种剂量的AZD 6482(KIN-193)以2倍稀释步骤使用,范围为5.12μM至0.02μM。使用PipelinePilot [1]中实施的固定顶部和底部S形拟合算法,确定与对照(未处理)孔相比50%细胞数的IC 50。 |
| 数据来源文献 | [1]. Ni J, et al. Functional characterization of an isoform-selective inhibitor of PI3K-p110β as a potential anticancer agent. Cancer Discov. 2012 May;2(5):425-33 |
| 规格 | 5mg 10mg 50mg |
AZD 6482 是一种有效的选择性 p110β 抑制剂。