Dinaciclib/SCH727965
货号:
ID1000
品牌:
Jinpan
暂无详情
产品简介
| EC | EINECS 214-589-6 |
| 别名 | SCH727965 |
| 英文名称 | Dinaciclib/SCH727965 |
| CAS | 779353-01-4 |
| 分子式 | C21H28N6O2 |
| 分子量 | 396.49 |
| 纯度 | ≥98% |
| 单位 | 瓶 |
| 生物活性 | Dinaciclib 是一种有效的选择性 CDK 抑制剂,抑制 CDK2,CDK5,CDK1 和 CDK9 的 IC50 分别为 1,1,3 和 4 nM[1-3]。 |
| IC50 | CDK1:3 nM ; CDK2:1 nM ; CDK5:1 nM ; CDK9:4 nM [1-3] |
| In Vitro | Dinaciclib(SCH 727965)是一种有效的DNA复制抑制剂,可阻断胸腺嘧啶(dThd)DNA在A2780细胞中的掺入,IC50为4 nM。 Dinaciclib(100 nM)抑制视网膜母细胞瘤(Rb)肿瘤抑制蛋白的磷酸化,并诱导p85 PARP caspase裂解产物的积累[1]。 Dinaciclib(SCH727965)以剂量依赖性方式抑制胰腺癌细胞的体外细胞生长。在与Dinaciclib孵育72小时后,对于MIAPaCa-2和Pa20C细胞,GI50分别为约10和20nM。这些结果与Dinaciclib在其他癌细胞系中的研究一致。在软琼脂测定中,5至10nM的Dinaciclib显著减少MIAPaCa-2细胞的集落形成和锚定独立生长。此外,使用BD FluoroChrom,改良的Boyden Chamber和伤口愈合测定[2]证明,通过Dinaciclib浓度从2-5nM开始,Pa20C和MIAPaCa-2细胞的体外细胞迁移显著降低。 |
| In Vivo | Dinaciclib(8,16,32和48mg/kg,ip)分别导致肿瘤抑制70%,70%,89%和96%; Dinaciclib(SCH 727965)耐受性良好,最高剂量组的最大体重减轻为5%。 Dinaciclib在小鼠中具有短的血浆半衰期。腹腔注射小鼠腹腔注射5 mg/kg Dinaciclib,血浆半衰期约为0.25小时[1]。使用Dinaciclib(SCH727965)进行治疗,每周两次ip剂量40mg/kg,持续4周,引起显著的肿瘤生长抑制(TGI)在10/10(100%)的低传代皮下胰腺异种移植物中测试[2]。 |
| 激酶实验 | 从Sf9细胞中纯化重组细胞周期蛋白/ CDK全酶,所述Sf9细胞经工程改造以产生表达特定细胞周期蛋白或CDK的杆状病毒。通常在含有50mM Tris-HCl(pH 8.0),10mM MgCl 2,1mM DTT和0.1mM原钒酸钠的激酶反应缓冲液中将细胞周期蛋白/ CDK复合物稀释至终浓度为50μg/ mL。对于每个激酶反应,将1μg酶和20μL2μM底物溶液(衍生自组蛋白H1的生物素化肽)混合并与10μL稀释的Dinaciclib(SCH727965)组合。通过添加50μL的2μMATP和0.1μCi的33P-ATP开始反应。将激酶反应在室温下温育1小时,并通过加入0.1%Triton X-100,1mM ATP,5mM EDTA和5mg/mL链霉抗生物素蛋白包被的SPA珠来终止。使用96孔GF/B滤板和Filtermate通用收获机捕获SPA珠。珠子用2M NaCl洗涤两次,用含有1%磷酸的2M NaCl洗涤两次。然后使用TopCount 96孔液体闪烁计数器测定信号。剂量 – 反应曲线由抑制性化合物的一式八份连续稀释产生。 IC50值通过非线性回归分析得出[1]。 |
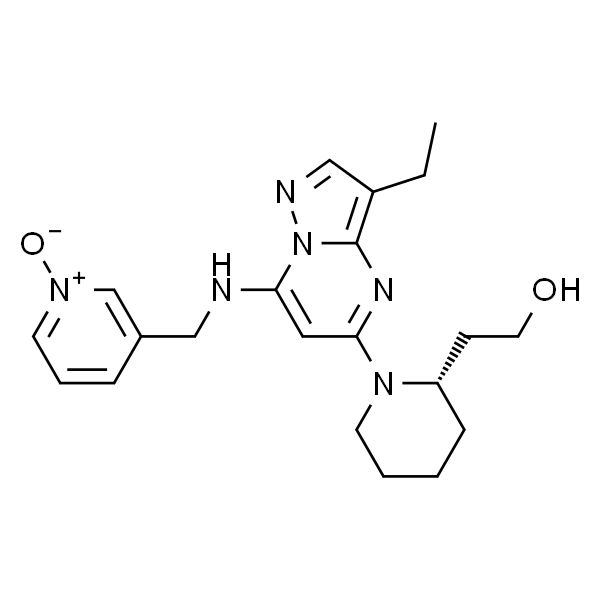
| SMILES | OCC[C@H]1N(CCCC1)C2=NC3=C(C=NN3C(NCC4=C[N+]([O-])=CC=C4)=C2)CC |
| 靶点 | CDK |
| 动物实验 | 小鼠[1]对于肿瘤植入,在体外培养特定细胞系,用PBS洗涤一次,并重悬于PBS中的50%基质胶中至终浓度为4×107至5×10 7个细胞/毫升。在侧腹区域向裸鼠注射0.1mL该悬浮液sc。肿瘤长度(L),宽度(W)和身高(H)通过卡尺每周两次测量每只小鼠,然后使用公式(L×W×H)/ 2计算肿瘤体积。当肿瘤体积达到100mm 3时,将动物随机分配至治疗组(10只小鼠/组),并用Dinaciclib(每天8,16,32和48mg/kg,ip)或单独的化学治疗剂腹膜内注射治疗。给药时间表见表和图例。在治疗期间和之后测量肿瘤体积和体重。 |
| 细胞实验 | 将A2780细胞接种到组织培养皿上并用合适的生长培养基繁殖。将生长的培养物暴露于增加浓度的Dinaciclib(0.75,1.5,3.15,6.25,12.5,25和500nM)或媒介物对照,通常持续7天。除去培养基后,将细胞用50%甲醇/ 50%丙酮固定5分钟,并用0.2%结晶紫在2%乙醇中染色5分钟。染色后,用5至10mL水洗涤细胞。将染色的细胞溶解在1%脱氧胆酸中,并使用SOFTmax PRO 4.3读板仪在600nm下测量所得溶液的吸光度。将Dinaciclib处理的样品的吸光度绘制为载体处理的对照的吸光度的百分比,并将数据报告为相对于这些对照的IC 50值。对于悬浮细胞系,使用alamarBlue Cell Viability Assay试剂盒[1]获得细胞活力的评估。 |
| 数据来源文献 | [1]. Parry D, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010 Aug;9(8):2344-53. [2]. Feldmann G, et al. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol Ther. 2011 Oct 1;12(7):598-609. [3]. Feldmann G, et al. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol Ther. 2011 Oct 1;12(7):598-609. |
| 规格 | 5mg 10mg 50mg |
Dinaciclib 是一种有效的选择性 CDK 抑制剂。