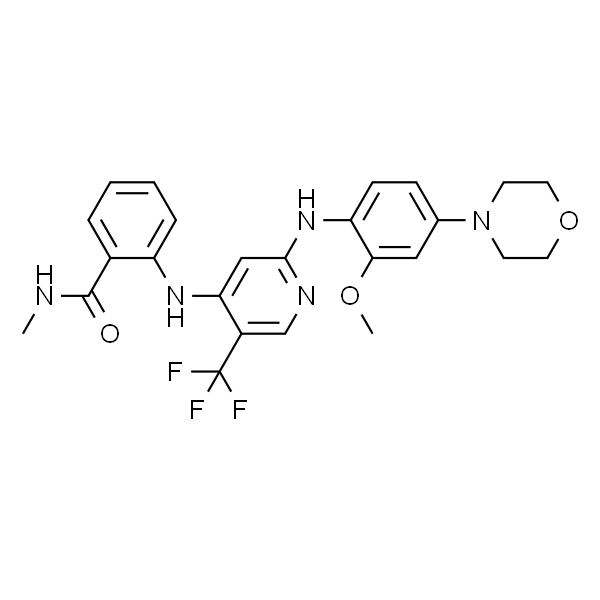
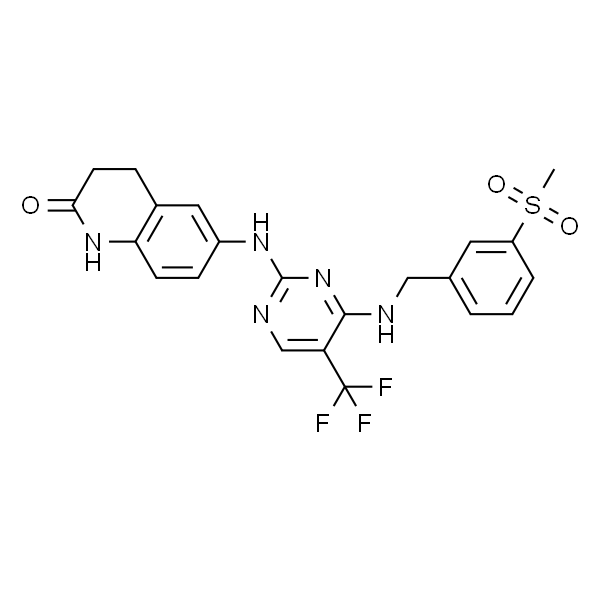
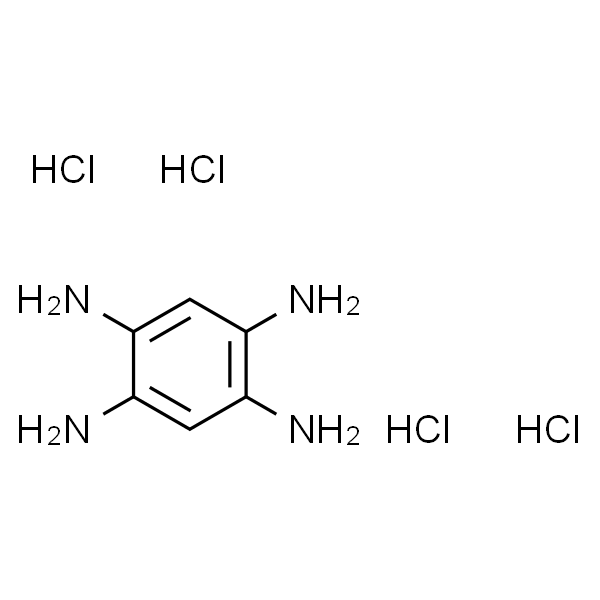
地法替尼
货号:
ID1280
品牌:
Jinpan
暂无详情
产品简介
| 别名 | VS-6063; PF-04554878 |
| 英文名称 | Defactinib |
| CAS | 1073154-85-4 |
| 分子式 | C20H21F3N8O3S |
| 分子量 | 510.49 |
| 储存条件 | 2-8°C |
| 纯度 | ≥98% |
| 单位 | 瓶 |
| 生物活性 | Defactinib 是一种新型 FAK 抑制剂,抑制 FAK 在 Tyr397 位点磷酸化,这种作用具有时间和剂量依赖性。[1] |
| In Vitro | VS-6063以时间和剂量依赖性方式抑制Tyr397位点的FAK磷酸化。 VS-6063和紫杉醇的组合显著降低增殖并增加细胞凋亡,导致肿瘤重量减少92.7%至97.9%。 RPPA数据显示VS-6063降低紫杉烷抗性细胞系中AKT和YB-1的水平。在所有细胞系中,VS-6063以剂量依赖性方式统计显著抑制pFAK(Tyr397)的表达。 VS-6063在3小时内抑制pFAK(Tyr397)的表达,48小时后逐渐恢复表达[1]。 |
| In Vivo | VS-6063剂量为25mg / kg,每天两次或更高,在3小时时统计学上显著抑制pFAK(Tyr397),24小时后表达恢复。因此,选择每天两次以25mg / kg施用VS-6063作为后续治疗实验的给药方案。对于治疗实验,将腹膜腔内携带HeyA8肿瘤的雌性裸鼠随机分成4组(每组n = 10):1)每日口服两次,每周腹膜内注射磷酸盐缓冲盐水(对照); 2)VS-6063 25 mg / kg,每日口服两次; 3)每周腹膜内注射PTX; 4)VS-6063每天口服两次25mg / kg,每周腹膜内注射PTX。在HeyA8模型中通过PTX单一疗法使肿瘤重量减少87.4%,并且联合治疗导致最大的肿瘤重量减少,减少97.9%(与PTX相比P = 0.05)。在SKOV3ip1模型中,与PTX相比,联合组中观察到肿瘤重量减少92.7%(P <0.001)[1]。 |
| 靶点 | FAK |
| 动物实验 | 小鼠[1]为了确定VS-6063的抗肿瘤作用,腹膜内注射SKOV3ip1,SKOV3-TR,HeyA8和HeyA8-MDR细胞。在肿瘤细胞注射后一周,将小鼠随机分配到4组10只小鼠(对照组,单独的PTX,单独的VS-6063和具有VS-6063的PTX);在注射后3-4周开始治疗。每周腹腔注射2mg / kg(SKOV3ip1和SKOV3-TR)或2.5mg / kg(HeyA8和HeyA8-MDR)的PTX;每天口服两次25mg / kg的VS-6063。对照小鼠每周一次腹膜内接受HBSS,并且每天口服两次载体。每天监测小鼠的治疗副作用,并在第35天(SKOV3ip1或SKOV3-TR),第28天(HeyA8或HeyA8-MDR)或当任何小鼠看起来垂死时被杀死。记录总体重,肿瘤发生率和质量以及肿瘤结节的数量。将肿瘤固定在福尔马林中或嵌入石蜡中或在液氮中以最佳切割温度(OCT)化合物快速冷冻。 |
| 数据来源文献 | [1]. Kang Y, et al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J Natl Cancer Inst. 2013 Oct 2;105(19):1485-95 |
| 规格 | 5mg 10mg 50mg |
是一种选择性的FAK抑制剂。Phase 2。