TMP269
货号:
IT1810
品牌:
Jinpan
暂无详情
产品简介
| 有效期 | 2年 |
| 英文名称 | TMP269 |
| CAS | 1314890-29-3 |
| 分子式 | C25H21F3N4O3S |
| 分子量 | 514.52 |
| 储存条件 | 2-8℃ |
| 纯度 | ≥98% |
| 外观(性状) | Solid |
| 单位 | 瓶 |
| 生物活性 | TMP269 是一种有效的,选择性的脱乙酰化酶 (HDAC) 抑制剂,能够抑制 HDAC 4/HDAC 5/HDAC 7/HDAC 9,IC50 值分别为 157 nM,97 nM,43 nM 和 23 nM。 |
| In Vitro | TMP269对10μM的人CD4 + T细胞的线粒体活性和/或活力没有影响,可用作鉴定IIa类HDAC酶的内源底物的工具[1]。在IEC-18肠上皮细胞中,TMP269阻止细胞周期进程,DNA合成和响应G蛋白偶联受体/ PKD1激活诱导的增殖[2]。与HDAC4敲低一样,TMP269显著增强由CFZ诱导的MM细胞系中的细胞毒性,上调ATF4和CHOP并诱导细胞凋亡。 TMP269在MM细胞系中没有高度乙酰化组蛋白H3K9或α-微管蛋白,证实它对I类或IIb类HDAC没有抑制作用。以剂量依赖的方式,TPM269诱导的细胞毒性与caspase-8,-9,-3和PARP的切割相关,与细胞凋亡一致[3]。 |
| 激酶实验 | HDAC 选择性测试:剂量-反应研究以10个不同浓度在Reaction Biology Corp上进行,不同浓度通过最大终化合物浓度为100 μM的反应混合物三倍连续稀释得到。所有试验都基于相同的原理,如上所诉的HDAC9试验:乙酰化的或三氟乙酰化的赖氨酸残基在荧光肽底物中被HDAC脱乙酰化。HDAC1,HDAC2,HDAC3,HDAC6,HDAC10 和HDAC11被用作基于残基379-382 p53 (Arg-His-Lys-Lys(Ac))的底物。对于HDAC8,使用基于p53 (Arg-His-Lys(Ac)-Lys(Ac))的残基379–382,二乙酰化的多肽底物。HDAC4,HDAC5,HDAC7 和HDAC9试验使用IIa类HDAC特定荧光底物(Boc-Lys(三氟乙酰基)-AMC)。所有反应与50 μM HDAC底物在包含1% DMSO终浓度的实验缓冲液(50 mM Tris-HCl,pH 8.0,137 mM NaCl,2.7 mM KCl,1 mM MgCl2,1 毫克/毫升BSA)中进行,与曲古抑菌素A和胰蛋白酶在30℃下培养2小时。[1] |
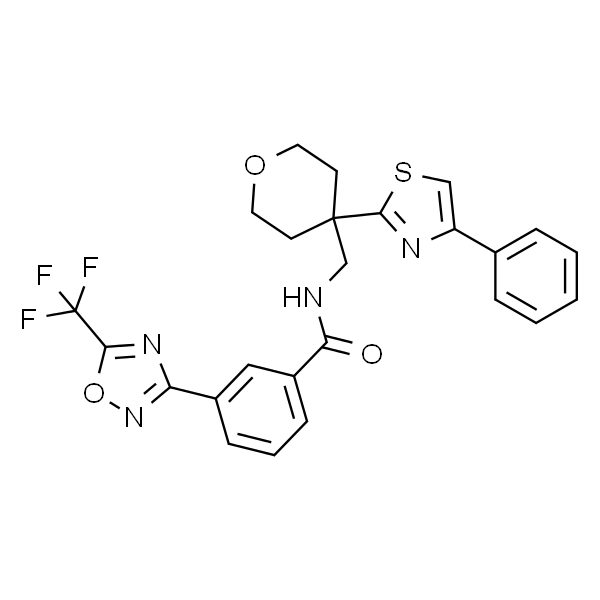
| SMILES | O=C(NCC1(C2=NC(C3=CC=CC=C3)=CS2)CCOCC1)C4=CC=CC(C5=NOC(C(F)(F)F)=N5)=C4 |
| 靶点 | HDAC |
| 数据来源文献 | [1]. Lobera M, et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol. 2013 May;9(5):319-25. [2]. Sinnett-Smith J, et al. Protein kinase D1 mediates class IIa histone deacetylase phosphorylation and nuclear extrusion in intestinal epithelial cells: role in mitogenic signaling. Am J Physiol Cell Physiol. 2014 May 15;306(10):C961-71. [3]. Kikuchi S, et al. Class IIa HDAC inhibition enhances ER stress-mediated cell death in multiple myeloma. Leukemia. 2015 Sep;29(9):1918-1927 |
| 规格 | 5mg 10mg |
TMP269 是强效的选择性的IIa类HDAC抑制剂。