GSK2801
货号:
IG2240
品牌:
Jinpan
暂无详情
产品简介
| 有效期 | 2年 |
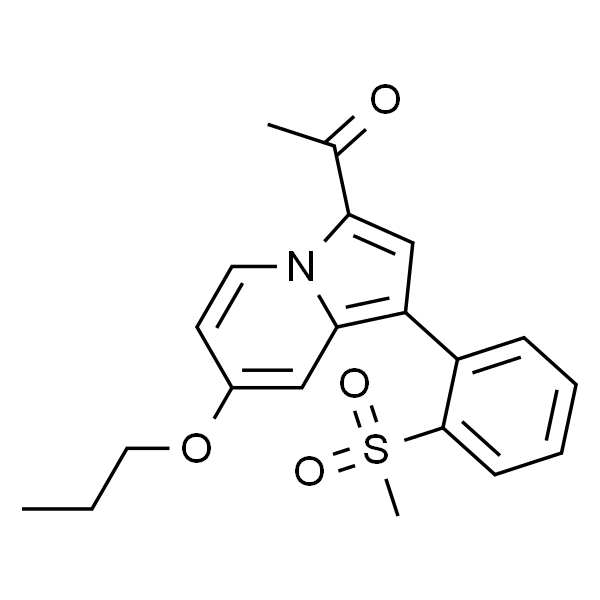
| 英文名称 | GSK2801 |
| CAS | 1619994-68-1 |
| 分子式 | C20H21NO4S |
| 分子量 | 371.45 |
| 储存条件 | -20℃ |
| 纯度 | ≥98% |
| 外观(性状) | Light yellow to khaki Solid |
| 单位 | 瓶 |
| SMILES | O=C(C)C1=CC(C2=C(S(=O)(C)=O)C=CC=C2)=C3N1C=CC(OCCC)=C3 |
| 靶点 | Epigenetic Reader Domain(BAZ2B,BAZ2A) |
| 规格 | 5mg 10mg |
GSK2801是一种选择性溴结构域 BAZ2A/B 抑制剂。