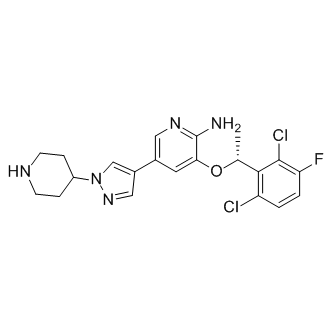
Crizotinib/PF-02341066;克唑替尼
货号:
IC1760
品牌:
Jinpan

暂无详情
产品简介
| EC | EINECS 638-814-9 |
| MDL | MFCD12407409 |
| 别名 | 可唑替尼;Xalkori;PF-2341066 |
| 英文名称 | Crizotinib/PF-02341066 |
| CAS | 877399-52-5 |
| 分子式 | C21H22Cl2FN5O |
| 分子量 | 450.34 |
| 纯度 | HPLC≥98% |
| 单位 | 瓶 |
| 生物活性 | Crizotinib是有效的 c-Met 和 ALK 抑制剂,在细胞试验中的 IC50 值分别为11 nM 和 24 nM。[1-8] |
| IC50 | 11 nM (c-Met), 24 nM (ALK)[1-8] |
| In Vitro | Crizotinib(PF-2341066)在mIMCD3小鼠或MDCK犬上皮细胞中显示出相似的针对c-Met磷酸化的效力,IC50分别为5nM和20nM。与NIH3T3细胞相比,Crizotinib(PF-2341066)显示出针对NIH3T3细胞的改善或相似的活性,所述NIH3T3细胞经工程改造以表达c-Met ATP结合位点突变体V1092I或H1094R或P-环突变体M1250T,IC50分别为19nM,2nM和15nM。表达野生型受体,IC50为13 nM。相反,与野生型受体相比,观察到针对经工程改造以表达c-Met活化环突变体Y1230C和Y1235D的细胞的Crizotinib(PF-2341066)效力的显著变化,IC50分别为127nM和92nM。 Crizotinib(PF-2341066)还有效地阻止了NCI-H69和HOP92细胞中c-Met的磷酸化,IC50分别为13 nM和16 nM,分别表达内源性c-Met变体R988C和T1010I [1]。 Crizotinib(PF-2341066)还有效抑制Karpas299或SU-DHL-1 ALCL细胞中的NPM-ALK磷酸化,IC50为24 nM。 Crizotinib(PF-2341066)有效阻止细胞增殖,这与ALK阳性ALCL细胞中G(1)-S期细胞周期停滞和诱导细胞凋亡有关,IC50为30 nM,而ALK阴性淋巴瘤细胞则不然[2]。此外,Crizotinib(PF-2341066)可预防与原发肿瘤生长(即增殖和存活)以及转移相关的骨肉瘤行为[3]。 |
| In Vivo | Crizotinib(PF-2341066)显示在50 mg/kg /天和75 mg/kg /天治疗队列中引起大规模肿瘤(> 600 mm3)明显消退的能力,平均肿瘤体积减少60%。 GTL-16模型中的日常管理计划。在另一项研究中,Crizotinib(PF-2341066)显示出完全抑制GTL-16肿瘤生长> 3个月的能力,12个小鼠中只有1只在3个月的治疗方案中显示出50mg/kg /的肿瘤生长显著增加。天。在GTL-16肿瘤中观察到12.5mg/kg /天,25mg/kg /天和50mg/kg /天的CD31阳性内皮细胞的显著剂量依赖性减少,表明MVD的抑制显示剂量与抗肿瘤疗效的相关性。 Crizotinib(PF-2341066)在GTL-16和U87MG模型中显示出人类VEGFA和IL-8血浆水平的显著剂量依赖性降低。 po给予Crizotinib(PF-2341066)后,在GTL-16肿瘤中观察到磷酸化c-Met,Akt,Erk,PLCλ1和STAT5水平的显著抑制[1]。 Crizotinib(PF-2341066)可预防与原发性肿瘤生长和转移相关的骨肉瘤行为。在通过口服强饲法用Crizotinib(PF-2341066)处理的裸鼠中,Crizotinib(PF-2341066)预防了骨肉瘤异种移植物的生长和相关的骨溶解和皮层外骨基质的形成[3]。用50mg/kg Crizotinib(PF-2341066)处理c-MET-扩增的GTL-16异种移植物引起肿瘤消退,其与18F-FDG摄取的缓慢减少和降低葡萄糖转运蛋白1,GLUT-1的表达相关[4]。 |
| 激酶实验 | 在用Crizotinib(PF-2341066)孵育细胞1小时和/或适当的配体指定时间后,用补充有1mM Na3VO4的HBSS洗涤细胞一次,并从细胞产生蛋白质裂解物。随后,通过夹心ELISA方法评估所选蛋白激酶的磷酸化,使用用于包被96孔板的特异性捕获抗体和对磷酸化酪氨酸残基特异的检测抗体。 |
| SMILES | ClC1=C(F)C=CC(Cl)=C1[C@H](OC2=CC(C3=CN(N=C3)C4CCNCC4)=CN=C2N)C |
| 靶点 | ALK;c-Met/HGFR;ROS1��ROS proto-oncogene 1 , receptor tyrosine kinase�� |
| 动物实验 | 在施用Crizotinib(PF-2341066)后的指定时间,对小鼠进行人道安乐死,并切除肿瘤。将肿瘤快速冷冻并使用液氮冷却的cryomortar和研杵进行粉碎,产生蛋白质裂解物,并使用BSA测定法测定蛋白质浓度。使用捕获ELISA或免疫沉淀 – 免疫印迹方法测定总蛋白和磷酸化蛋白的水平。 |
| 细胞实验 | 在补充有10%FBS(生长培养基)的培养基中以低密度将肿瘤细胞接种在96孔板中,24小时后转移至无血清培养基(0%FBS和0.04%BSA)。向每个孔中加入适当的对照或指定浓度的Crizotinib(PF-2341066),并将细胞温育24至72小时。将人脐带血管内皮细胞(HUVEC)以每孔> 20,000个细胞接种于EGM2培养基中的96孔板中5至6小时,并转移至无血清培养基中过夜。第二天,向每个孔中加入适当的对照或指定浓度的Crizotinib(PF-2341066),并在孵育1小时后,将HGF以100ng/mL加入指定的孔中。进行测定以确定相对肿瘤细胞或HUVEC数。 |
| 数据来源文献 | [1]. Zou HY, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007, 67(9), 4408-4417. [2]. Christensen JG, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007, 6(12 Pt 1), 3314-3322. [3]. Sampson ER, et al. The orally bioavailable met inhibitor PF-2341066 inhibits osteosarcoma growth and osteolysis/matrix production in a xenograft model. J Bone Miner Res. 2011, 26(6), 1283-1294. [4]. Cullinane C, et al. Differential (18)F-FDG and 3′-deoxy-3′-(18)F-fluorothymidine PET responses to pharmacologic inhibition of the c-MET receptor in preclinical tumor models. J Nucl Med. 2011 Aug;52(8):1261-7 [5]. Shen A, et al. c-Myc alterations confer therapeutic response and acquired resistance to c-Met inhibitors in MET-addicted cancers. Cancer Res. 2015 Nov 1;75(21):4548-59. [6]. Umapathy G, et al. The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Sci Signal. 2014 Oct 28;7(349):ra102. [7]. Tucker ER, et al. Immunoassays for the quantification of ALK and phosphorylated ALK support the evaluation of on-target ALK inhibitors in neuroblastoma. Mol Oncol. 2017 Aug;11(8):996-1006. [8]. Liu H, et al. Identifying and Targeting Sporadic Oncogenic GeneticLiu H, et al. Identifying and Targeting Sporadic Oncogenic Genetic Aberrations in Mouse Models of Triple Negative Breast Cancer. Cancer Discov. 2018 Mar;8(3):354-369. |
| 规格 | 5mg 10mg 20mg 50mg |
Crizotinib是一种有效的c-Met和ALK抑制剂,也是 ROS1 抑制剂。Crizotinib具有有效的肿瘤生长抑制作用,可用于相关研究。