| 有效期 |
2年 |
| 别名 |
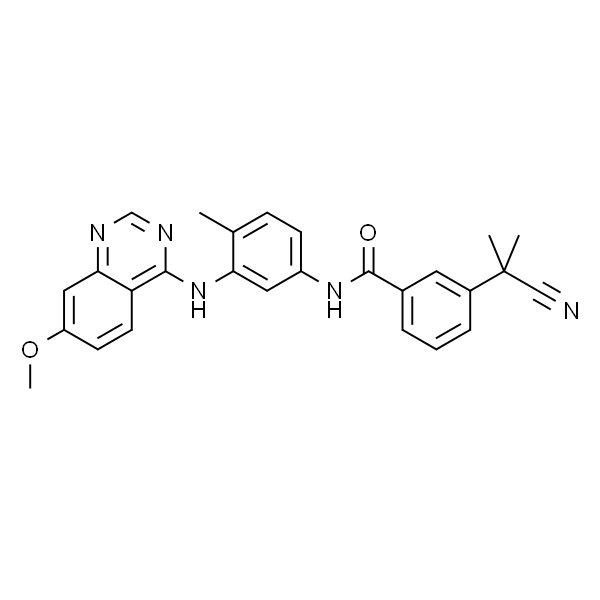
3-(2-氰基丙烷-2-基)-N-(3-((7-甲氧基-4-基)氨基)-4-甲基苯基)苯甲酰胺 |
| 英文名称 |
AZ-304 |
| CAS |
942507-42-8 |
| 分子式 |
C27H25N5O2 |
| 分子量 |
451.52 |
| 储存条件 |
2-8℃ |
| 纯度 |
≥98% |
| 外观(性状) |
White to off-white (Solid) |
| 单位 |
瓶 |
| 生物活性 |
AZ304 is an ATP-competitive dual BRAF kinase inhibitor, potently inhibits wild type BRAF, V600E mutant BRAF and wild type CRAF, with IC50s of 79 nM, 38 nM and 68 nM, respectively. AZ304 also has significant effect on other kinases, such as p38 (IC50, 6 nM), CSF1R (IC50, 35 nM). Anti-tumor activity[1]. |
| In Vitro |
AZ304 (1 nM-100 μM) potently reduces ERK phosphorylation (p-ERK), with a mean EC50 of 65 nM in the V600E mutant BRAF containing melanoma cell line A375, and EC50s of 52 nM, 60 nM in the wild type BRAF melanoma cell line SK-MEL-31 with and without EGF[1]. AZ304 also potently inhibits p-p38 in both BRAF genetic statuses cell lines[1].AZ304 (2 μM, 36 and 48 hours) decreases BRAF, p-ERK, p-AKT and p-mTOR levels, increases p-EGFR in both BRAF V600E mutant and BRAF wild type cells. AZ304 down-regulates p-EGFR, inhibits p-ERK, more potently suppresses BRAF, ERK, AKT and mTOR signalling pathways in combination with C225[1]. |
| In Vivo |
Compared with vehicle-treated controls, treatment with AZ304 or Cetuximab alone resulted in reduced tumor growth in both xenograft models. Furthermore, the AZ304 and Cetuximab combination caused dramatic tumour growth inhibition and even shrinking in the Caco-2 xenograft model.[1] |
| 激酶实验 |
Briefly, the kinase activity of BRAF WT, BRAF V600E, or CRAF was measured using an AlphaScreen assay monitoring MEK1/2 phosphorylation at Ser217/221. For CDK2 and CDK4 kinases, activity was also measured by AlphaScreen, monitoring phosphorylation of biotin Rb peptide at Ser780. Similarly, MAP3K7, CSF1R, and JAK2 kinase activity were measured by phosphorylation of their biotinylated substrates MKK6 kinase-dead protein at Ser271/Thr275, or tyrosine phosphorylation of pEY or Tyk2 Tyr1054/1055 peptides, respectively. CSK, IGF1R, EGFR, FGFR1 and SRC kinase activity was measured using a sandwich ELISA detecting phosphorylated poly EAY peptide with a HRP conjugated phosphotyrosine antibody and TMB substrate, while p38 kinase activity was measured by monitoring phosphorylation of MBP protein with radiolabeled 33P-ATP in a filter binding format. All assays were screened under respective ATP Km conditions and inhibitor IC50s were derived from either 5 (RAF kinases, CDK2, CDK4, MAP3K7, JAK2), 10 (CSK, IGF1R, EGFR, FGFR1, SRC) or 11 (p38, CSF1R) point compound dose response.[1] |
| SMILES |
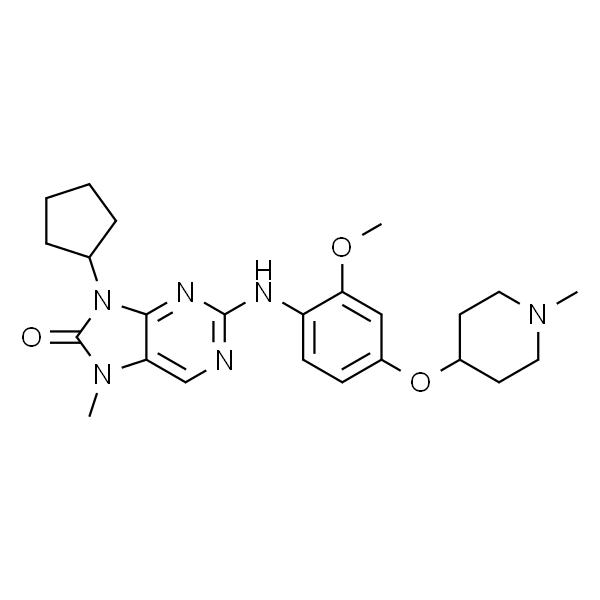
O=C(NC1=CC=C(C)C(NC2=C3C=CC(OC)=CC3=NC=N2)=C1)C4=CC=CC(C(C)(C#N)C)=C4 |
| 靶点 |
p38,CSF1R,C-Raf,B-Raf |
| 动物实验 |
AZ304 (0, 0.1, 1, 10, 100 μM, 48 and 72 hours) dose-dependently inhibits the growth of RKO, HT-29, DiFi, and Caco-2, with GI50s of 4.539 μM, 3.896 μM, 4.987 μM, 1.763 μM (48 hours) and 0.5032 μM, 0.3887 μM, 0.6354 μM, 0.3772 μM (72 hours), respectively[1]. |
| 细胞实验 |
Female 4–6 weeks old athymic BALB/c nude mice were purchased from Shanghai SLAC Laboratory Animal Centre. RKO/Caco-2 cells (1 × 10^7) in 200 μl PBS were injected subcutaneously into the right scapular region of mice. After the average tumour size reached 150 – 200 mm^3, animals were randomly divided into 4 groups, each containing three mice and were treated with vehicle only (CON) which orally received 0.5% HPMC and injected with 0.9% saline, AZ304 only (AZ304 dissolved in 0.5% HPMC, 10 mg/kg by oral gavage twice daily), Cetuximab only (40 mg/kg by intraperitoneal injection twice per week), or their combination (A+C) for 10 days. Tumors were measured with a caliper every 2 days, so did body weights. Tumor volume was calculated using the formula V = 1/2 (width^2 × length). Mice were terminated by CO2 inhalation when the tumor diameters reached 1.5 cm, according to the protocol filed with the Guidance of Institutional Animal Care and Use Committee of China Medical University.[1] |
| 数据来源文献 |
[1]. Ma R, et al. AZ304, a novel dual BRAF inhibitor, exerts anti-tumour effects in colorectal cancer independently of BRAF genetic status. Br J Cancer. 2018 May;118(11):1453-1463. |
| 规格 |
10mg 25mg |