JQ1

| 含量 | Purity≥98% |
| 别名 | (+)-JQ1 |
| CAS | 1268524-70-4 |
| 分子式 | C23H25ClN4O2S |
| 分子量 | 456.99 |
| 纯度 | ≥98% |
| 单位 | 支 |
| 生物活性 | (+) -JQ-1 是一种 BET bromodomain 抑制剂, 抑制 BRD4 (1/2) 的 IC50 分别为 77 nM 和 33 nM[1]。 |
| IC50 | IC50: 77/33 nM (BRD4 (1/2) ) [1] |
| In Vitro | (+) – JQ1代表溴结构域BET家族的有效, 高度特异性和Kac竞争性抑制剂。 (+) – JQ1 (100nM, 48小时) 促进细胞纺锤, 扁平化和角蛋白表达增加所表现出的鳞状分化。 (+) – JQ1 (250 nM) 诱导处理的NMC 797细胞中角蛋白的快速表达, 与 ( – ) – JQ1 (250 nM) 和载体对照相比, 通过定量免疫组织化学测定。 (+) – JQ1 (250 nM) 引发与 ( – ) – JQ1 (250 nM) 相比, 处理的NMC 797细胞的强 (3+) 角蛋白染色的时间依赖性诱导[1]。 (+) – JQ1是BET家族共激活蛋白BRD4的有效噻东嗪抑制剂 (Kd = 90nM) , 其通过MYC致癌基因的转录控制参与癌症的发病机理。 (+) – JQ1的剂量范围研究表明H4Kac4结合的有效抑制, 小鼠BRDT (1) 的IC50值为10nM, 人BRDT (1) 的IC50值为11nM [2]。 |
| In Vivo | 将具有已建立的肿瘤的匹配的小鼠组随机化为用 (+) – JQ1 (50mg / kg) 或载体治疗, 通过每日腹膜内注射施用。在随机化之前和治疗四天后, 通过FDG-PET成像评估小鼠。用 (+) – JQ1处理观察到FDG摄取显着降低。肿瘤体积测量证实JQ1治疗可减少肿瘤生长。 (+) – JQ1的药代动力学研究在静脉内和口服给药后在CD1小鼠中进行。静脉内给药 (5mg / kg) 后 (+) – JQ1的平均血浆浓度 – 时间曲线。静脉注射 (+) – JQ1的药代动力学参数表现出优异的药物暴露 (AUC = 2090hr * ng / mL) 和约1小时的半衰期 (T1 / 2) 。口服给药后 (+) – JQ1的平均血浆浓度 – 时间曲线 (10mg / kg) 。口服 (+) – JQ1的药代动力学参数显示出优异的口服生物利用度 (F = 49%) , 峰值血浆浓度 (Cmax = 1180ng / mL) 和药物暴露 (AUC = 2090hr * ng / mL) [1]。 |
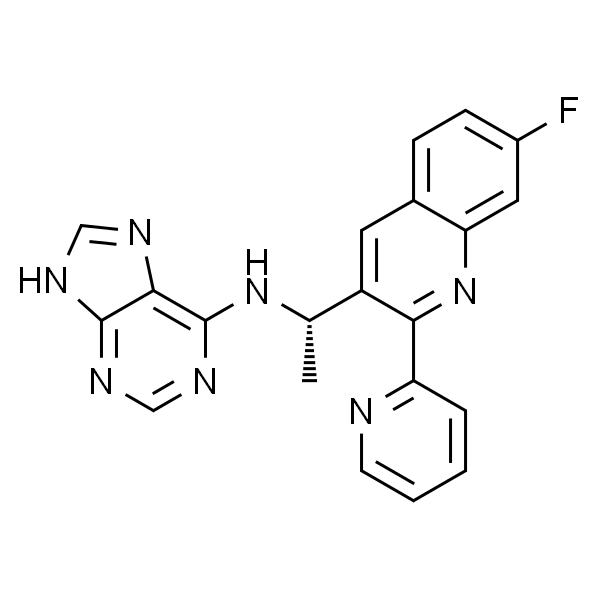
| SMILES | O=C(C[C@H]1C2=NN=C(N2C3=C(C(C4=CC=C(C=C4)Cl)=N1)C(C)=C(S3)C)C)OC(C)(C)C |
| 靶点 | Epigenetic Reader Domain |
| 动物实验 | 小鼠[1]将具有确定肿瘤的小鼠的匹配群组随机化至用 (+) – JQ1 (50mg / kg) 或载体治疗, 通过每日腹膜内注射施用。对于静脉内尾静脉注射研究, 用5mg / kg的单剂量 (+) – JQ1处理雄性CD1小鼠 (24-29g) , 对于口服管饲研究, 用10mg / kg处理雄性CD1小鼠 (24-29g) 。在麻醉下用异氟醚以预定的时间间隔 (0.033, 0.083, 0.25, 0.5, 1, 2, 4, 5, 8和24小时) 通过眼眶后穿刺从动物中取出约150μL血液。每个时间点分析三只动物。将血液样品置于冰上并离心以在取样后15分钟内获得血浆样品 (2000×g, 4℃下5分钟) 。将血浆样品储存在约-70℃直至进行分析。在整个研究过程中, 小鼠可以免费获得食物和水。大鼠[2]用载体或 (+) – JQ1 (10mg / kg) 处理成年雄性Sprague-Dawley大鼠。以1/100体重IP治疗。每天两次检查大鼠的死亡率并在第1, 3, 7, 14和21天称重。治疗方案使用每天施用50mg / kg JQ1的4天, 其余每天两次降至10mg / kg。该研究由于在一部分动物中出现不良反应。对于完成3周治疗的所有动物, 如小鼠研究所述测定睾丸质量, 精子活力和精子计数。简而言之, 睾丸固定在Bouin’s并为组织学做准备。另一半用温热的M16缓冲液切碎, 用于精子计数和运动性研究。 |
| 细胞实验 | 将NUT中线癌患者细胞系 (797和11060) 接种在T-25烧瓶中, 并在含有10%胎牛血清和1%青霉素/链霉素的DMEM (797) 或RPMI (11060) 中生长。用250nM (+) – JQ1, 250nM ( – ) – JQ1或等体积的DMSO (0.025%) 处理细胞。在所需的时间点, 将2×10 6个细胞在4℃以500×g旋转5分钟并用PBS洗涤。将沉淀重悬于1mL冷PBS中并逐滴加入, 同时在15mL聚丙烯离心管中轻轻涡旋至9mL 70%乙醇。然后将固定的细胞在-20℃下冷冻过夜。第二天, 将细胞在4℃下以500×g离心10分钟, 并用3mL冷PBS洗涤。将细胞重悬于500μL碘化丙锭染色溶液 (0.2mg / mL RNAse A, 0.02mg / mL碘化丙啶, 0.1%Triton-X的PBS溶液) 中, 并在37℃下孵育20分钟。然后将样品转移到冰上并在BD FACS Canto II上分析。生成直方图并使用FlowJo流式细胞术分析软件[1]进行细胞周期分析。 |
| 数据来源文献 | [1]. Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010 Dec 23; 468 (7327) :1067-73.
[2]. Matzuk MM, et al. Small-molecule inhibition of BRDT for male contraception. Cell. 2012 Aug 17; 150 (4) :673-84. [3]. Peirs S, et al. Targeting BET proteins improves the therapeutic efficacy of BCL-2 inhibition in T-cell acute lymphoblastic leukemia. Leukemia. 2017 Feb 3. [4]. T?gel L, et al. Dual Targeting of Bromodomain and Extraterminal Domain Proteins, and WNT or MAPK Signaling, Inhibits c-MYC Expression and Proliferation of Colorectal Cancer Cells. Mol Cancer Ther. 2016 Jun; 15 (6) :1217-26. [5]. Sahni JM, et al. Bromodomain and Extraterminal Protein Inhibition Blocks Growth of Triple-negative Breast Cancers through the Suppression of Aurora Kinases. J Biol Chem. 2016 Nov 4; 291 (45) :23756-23768. [6]. Nakamura Y, et al. Targeting of super-enhancers and mutant BRAF can suppress growth of BRAF-mutant colon cancer cells via repression of MAPK signaling pathway. Cancer Lett. 2017 Aug 28; 402:100-109. [7]. Bhattacharyya S, et al. Altered hydroxymethylation is seen at regulatory regions in pancreatic cancer and regulates oncogenic pathways. Genome Res. 2017 Nov; 27 (11) :1830-1842. [8]. Lv B, et al. Enhancement of adenovirus infection and adenoviral vector-mediated gene delivery by bromodomain inhibitor JQ1. Sci Rep. 2018 Aug 1; 8 (1) :11554. doi: 10.1038/s41598-018-28421-x. [9]. Huang X, et al. Targeting Epigenetic Crosstalk as a Therapeutic Strategy for EZH2-Aberrant Solid Tumors. Cell. 2018 Sep 20; 175 (1) :186-199.e19. doi: 10.1016/j.cell.2018.08.058. Epub 2018 Sep 13. |
| 备注 | 以上数据均来自公开文献, Jinpan暂未进行独立验证, 仅供参考。These protocols are for reference only. Jinpan does not independently validate these methods. |
| 规格 | 5mg 10mg 25mg |
(+)-JQ-1 是一种 BET bromodomain 抑制剂。