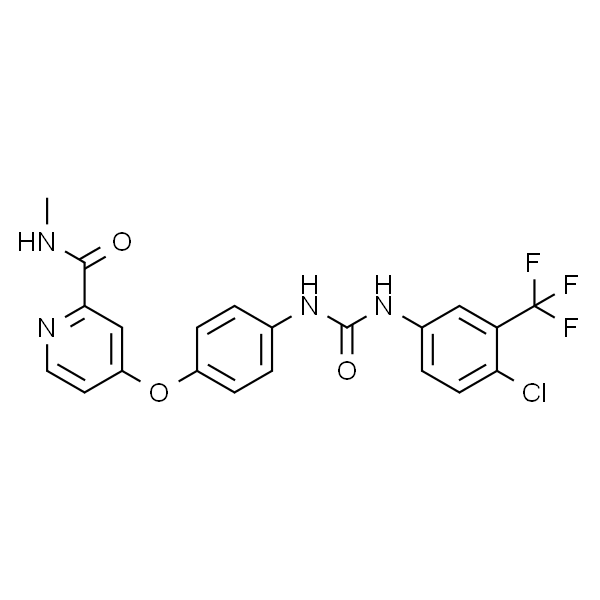
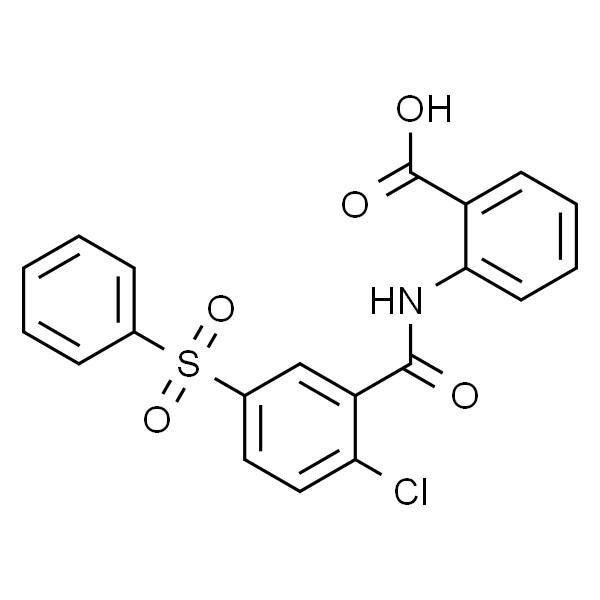
BAY-8002
货号:
IB1430
品牌:
Jinpan
暂无详情
产品简介
| 有效期 | 2年 |
| 描述 | 是一种有效、选择性的单羧酸转运蛋白第 1 亚型 (monocarboxylate transporter 1 (MCT1)) 抑制剂。 |
| MDL | MFCD05157149 |
| InChIKey | CLAUJSRBKSRTGQ-UHFFFAOYSA-N |
| InChI | InChI=1S/C20H14ClNO5S/c21-17-11-10-14(28(26,27)13-6-2-1-3-7-13)12-16(17)19(23)22-18-9-5-4-8-15(18)20(24)25/h1-12H,(H,22,23)(H,24,25) |
| PubChem CID | 992293 |
| CAS | 724440-27-1 |
| 分子式 | C20H14ClNO5S |
| 分子量 | 415.85 |
| 储存条件 | -20℃ |
| 纯度 | ≥98% |
| 外观(性状) | Solid |
| 单位 | 瓶 |
| SMILES | O=C(O)C1=CC=CC=C1NC(C2=CC(S(=O)(C3=CC=CC=C3)=O)=CC=C2Cl)=O |
| 靶点 | Monocarboxylate Transporter(MCT) |
| 规格 | 5mg |