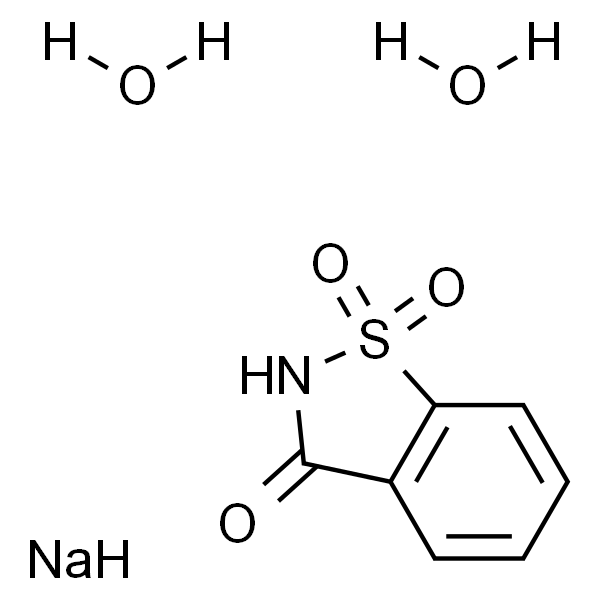
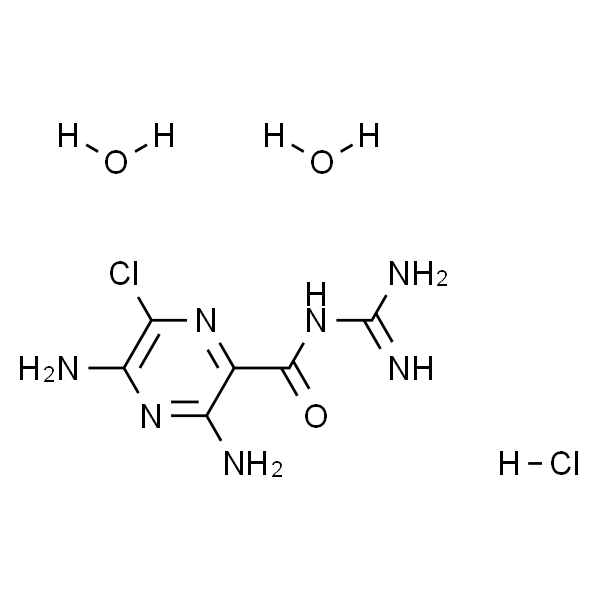
Amiloride hydrochloride dihydrate (MK-870 hydrochloride dihydrate) is an inhibitor of both epithelial sodium channel (ENaC) and urokinase-type plasminogen activator receptor (uTPA). Amiloride hydrochloride dihydrate is a blocker of polycystin-2 (PC2; TRPP2) channel.[1-8]
Amiloride hydrochloride dihydrate is a potent epithelial sodium channels (ENaCs) blocker. Amiloride is highly concentrated in the plasma 15 to 30 minutes following the injections. 2 mg/kg dose of Amiloride hydrochloride dihydrate has no effect on blood pressure, heart rate, mesenteric vascular resistance, or hindquarters vascular resistance as compare to the baseline measurements (n=7). Over a 2 hour period, Amiloride hydrochloride dihydrate elicits negligible responses in arterial pressure (-1±1 mmHg) and heart rate (-10±6 bpm/min) as compare to baseline levels. Results show an Amiloride hydrochloride dihydrate dose-related response pattern for the c-Fos activation in the area postrema (AP). Even at the lowest dose of Amiloride hydrochloride dihydrate (0.1 mg/kg), the number of c-Fos labeled neurons in the AP is statistically different from the control rats at a p<0.01 level[6].
货期
1-2天
SMILES
O=C(C1=NC(Cl)=C(N)N=C1N)NC(N)=N.[H]Cl.O.O
靶点
Sodium Channel
动物实验
The experiments are performed on male and female Sprague-Dawley rats that have been maintained in a heat controlled room maintained at 23° C with a 12h/12h light-dark schedule. A total of 116 adult rats are injected i.p. with Amiloride hydrochloride dihydrate (0.1, 0.5, and 2.0 mg/kg). After 15, 30, 45, or 120 min post-injection, the rats are anesthetized with 8% chloral hydrate, the chest is opened, and blood samples are obtained by cardiac puncture. The blood is centrifuged at 5000 RPM for 5 min, and the plasma (1.5 mL) separated and stored at -80° C[6].
数据来源文献
[1] Kim KM, et al. Oncogene,?005, 24(3), 355-366. [2] Koivusalo M, et al. J Cell Biol,?010, 188(4), 547-563. [3] Karmazyn M, et al. Am J Physiol,?988, 255(3 Pt 2), H608-615. [4] Besterman JM, et al. J Biol Chem,?985, 260(2), 1155-1159. [5] Soltoff SP, et al. Science,?983, 220(4600), 957-958. [6]. Miller RL, et al. Blockade of ENaCs by amiloride induces c-Fos activation of the area postrema. Brain Res. 2015 Mar 19;1601:40-51. [7]. Xu LB, et al. Amiloride, a urokinase-type plasminogen activator receptor (uTPA) inhibitor, reduces proteinurea in podocytes. Genet Mol Res. 2015 Aug 14;14(3):9518-29. [8]. Giamarchi A, et al. A polycystin-2 (TRPP2) dimerization domain essential for the function of heteromeric polycystin complexes. EMBO J. 2010 Apr 7;29(7):1176-91.
For initial study of cellular internalization, NPs-EPI was incubated with tumor spheroids at an EPI concentration of 8 µg/mL for 4 h. The tumor spheroids were grown as our previous reported procedure. Briefly, HepG2 cells were seeded in 96-well plates at a density of 8×103 per well precoated with 50 µL of 1% low melting point agarose. Cells were cultured to obtain multicellular spheroids (400–500 μm in diameter) for subsequent studies. After incubation with NPs-EPI, tumor spheroids were rinsed three times with cold PBS and processed for image analysis by TEM.
To investigate the internalization pathways of NPs-EPI, HepG2 cells were seeded into 6-well plates with 2×105 cells per well and incubated for 24 hours. The cells were then pre-treated with different endocytic inhibitors (chlorpromazine hydrochloride 20 µg/mL, amiloride hydrochloride 200 µg/mL) at 37°C for 1 hour. Subsequently, NPs-EPI (EPI 8 µg/mL) were added and incubated at 37°C for 2 hours. Finally, the cells were washed three times with cold PBS and harvested for flow cytometry to determine the fluorescence intensity of EPI (Ex=488, Em=588). Cells pre-treated with PBS at 37°C served as the positive control group. Internalization assays were also performed in the absence of endocytic inhibitors at 4°C when indicated.
来源文献:Chen E, Han S, Song B, Xu L, Yuan H, Liang M, Sun Y. Mechanism Investigation of Hyaluronidase-Combined Multistage Nanoparticles for Solid Tumor Penetration and Antitumor Effect. Int J Nanomedicine. 2020 Aug 24;15:6311-6324. doi: 10.2147/IJN.S257164. PMID: 32922003; PMCID: PMC7458542.
Alvimopan dihydrate (ADL 8-2698 dihydrate) is a potent, selective, orally active and reversible μ-opioid receptor antagonist, with an IC50 of 1.7 nM. Alvimopan dihydrate has selectivity for μ-opioid receptor (Ki=0.47 nM) over κ- and δ-opioid receptors (Kis=100, 12 nM, respectively). Alvimopan dihydrate can be used for the research of postoperative ileus[1-[3].
IC50
1.7 nM (μ-opioid receptor)[1]
In Vitro
Alvimopan inhibits the loperamide-stimulated [35S]GTPγS binding to membranes containing the cloned human μ-opioid receptor, with an IC50 of 1.7 nM[1].
In Vivo
Alvimopan (0.1-1.0 mg/kg; p.o.) partially antagonizes the slowing of small intestinal transit of 113Sn-labelled microspheres produced by morphine in rats.Alvimopan (3 mg/kg; p.o.) has no effect on the visceromotor behavioural responses (VMR) induced by noxious colorectal distension (CRD) in conscious rats[3].