| MDL |
MFCD16038900 |
| 别名 |
伊斯平斯 |
| 英文名称 |
PHA-680632 |
| CAS |
398493-79-3 |
| 分子式 |
C28H35N7O2 |
| 分子量 |
501.62 |
| 纯度 |
≥95% |
| 单位 |
瓶 |
| 生物活性 |
PHA-680632是极光激酶抑制剂 (aurora) , 对aurora A, B和C的 IC50 值分别为27, 135和120 nM[1-2]。 |
| IC50 |
Aurora A:27 nM (IC50)
Aurora B:135 nM (IC50)
Aurora C:120 nM (IC50) [1-2] |
| In Vitro |
与Aurora A相比, PHA-680632显示FLT3, LCK, PLK1, STLK2, VEGFR2和VEGFR3的IC50高30至200倍.PHA-680632在多种细胞类型中具有有效的抗增殖活性。对于C33A, HeLa, HCT116, HT29, LOVO, A549, MCF7, A2780, U2OS, DU145, IC50为0.32, 0.41, 0.06, 1.17, 0.56, 0.62, 0.29, 0.11, 1.56, 0.62, 0.07, 0.13, 0.41μM, U937, HL60, NHDF。 PHA-680632可在肿瘤细胞中引起多倍性。 PHA-680632细胞处理诱导类似于Aurora A或B耗尽的表型[1]。 PHA680632抑制不同癌细胞系中的集落形成并诱导多倍体。 PHA680632对Aurora-A的抑制增强了癌细胞的辐射反应, 尤其是p53缺陷细胞[2]。 |
| In Vivo |
PHA-680632抑制动物模型中的肿瘤生长。在HL60人急性髓性白血病异种移植模型中, 45mg / kg剂量的PHA-680632治疗导致85%的TGI没有毒性迹象。在A2780人卵巢癌模型中, PHA-680632以60mg / kg静脉内处理5天导致78%的TGI没有毒性迹象[1]。与辐射相关的PHA680632在癌细胞中产生累加效应, 特别是在p53缺陷细胞中, 但不起放射增敏剂的作用[2]。 |
| 激酶实验 |
使用闪烁亲近测定形式评估PHA-680632对激酶活性的抑制。在该测定中, 在存在用γ33-ATP追踪的ATP的情况下, 生物素化的底物被激酶转磷酸化。然后使用链霉抗生物素蛋白包被的闪烁亲近测定珠捕获磷酸化的底物, 并且在静置4小时后通过β-计数器评估磷酸化程度, 以使珠在密集的5M CsCl溶液上漂浮。特别地, 源自Chocktide序列的肽 (LRRWSLGL) 用作Aurora A的底物, 而优化的肽Auroratide1用于Aurora B和C.该测定在96孔板上以自动化形式进行。评估化合物对极光激酶和属于我们的激酶选择性筛选组的29种其他激酶的效力, 并确定相关的IC50 [1]。 |
| SMILES |
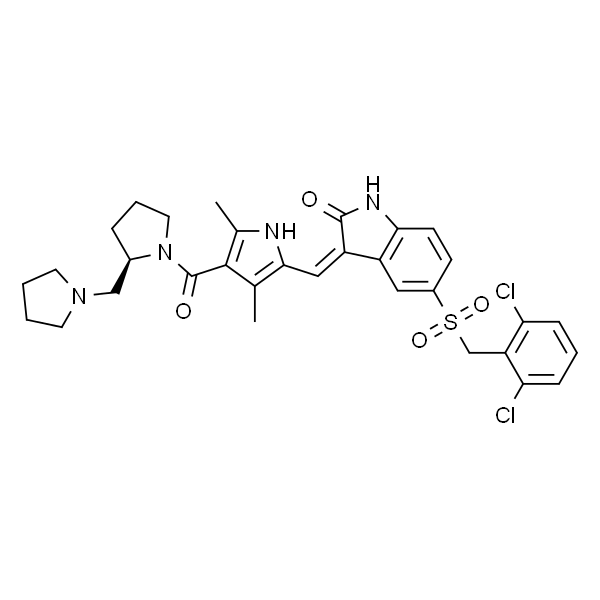
O=C(N1CC2=C(NN=C2NC(C3=CC=C(N4CCN(C)CC4)C=C3)=O)C1)NC5=C(C=CC=C5CC)CC |
| 靶点 |
Aurora Kinase |
| 动物实验 |
小鼠:将肿瘤异种移植小鼠随机分成四组 (每组六只小鼠) :A, 对照; B, 单独使用IR, 1天内8 Gy; C, PHA-680632单独, 40mg / kg, bid, 4天; D, 相同剂量的PHA-680632与IR组合 (第一次施用PHA680632后24小时, 与IR单独相似) , 持续4天。腹膜内 (ip) 施用药物或媒介物对照 (在5%葡萄糖溶液中相同体积的20%吐温-80) 。使用电子卡尺每周测量肿瘤大小两次。进行个体小鼠的随访。肿瘤体积由2D肿瘤测量估计[2]。 |
| 细胞实验 |
在24孔板中用适当的完全培养基以5, 000至15, 000cm 2的不同密度接种细胞。 24小时后, 用PHA-680632处理平板, 并在37℃, 5%CO 2气氛中温育72小时。在孵育时间结束时, 将细胞从每个平板上分离并使用细胞计数器计数。使用生长百分比与未治疗对照[1]计算IC50。 |
| 数据来源文献 |
[1]. Soncini C, et al. PHA-680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin Cancer Res. 2006 Jul 1; 12 (13) :4080-9.
[2]. Tao Y, et al. Enhancement of radiation response by inhibition of Aurora-A kinase using siRNA or a selective Aurora kinase inhibitor PHA680632 in p53-deficient cancer cells. Br J Cancer. 2007 Dec 17; 97 (12) :1664-72. |
| 备注 |
以上数据均来自公开文献, Jinpan暂未进行独立验证, 仅供参考。
These protocols are for reference only. Jinpan does not independently validate these methods. |
| 规格 |
5mg 10mg 50mg |