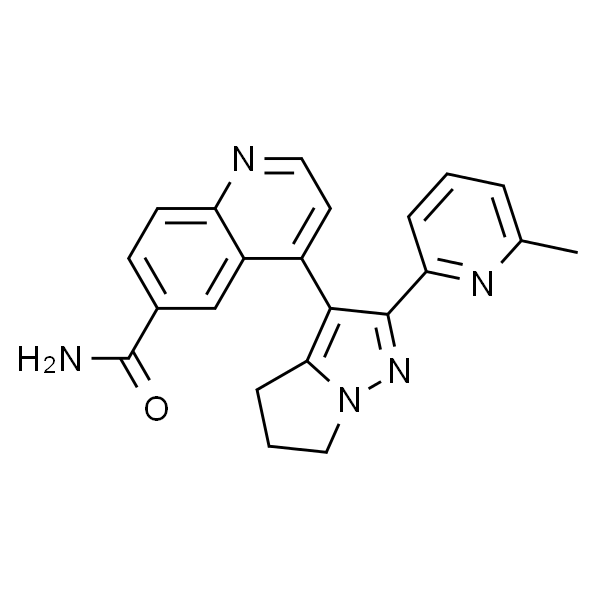
GSK343
货号:
IG0520
品牌:
Jinpan
暂无详情
产品简介
| MDL | MFCD00072464 |
| 别名 | 1-异丙基-N-[(6-甲基-2-氧代-4-丙基-1,2-二氢-3-吡啶基)甲基]-6-[2-(4-甲基-1-哌嗪基)-4-吡啶基]-1H-吲唑-4-甲酰胺 |
| CAS | 1346704-33-3 |
| 分子式 | C31H39N7O2 |
| 分子量 | 541.69 |
| 纯度 | ≥98% |
| 单位 | 瓶 |
| 生物活性 | GSK343 是一种高效,选择性的 EZH2 抑制剂,IC50 为 4 nM[1-2]。 |
| IC50 | 4 nM (EZH2), 240 nM (EZH1)[1] |
| In Vitro | GSK343在HeLa细胞中具有13μM的半数抑制浓度值,在SiHa细胞中具有15μM[2]。GSK343在吡啶酮的4-位含有正丙基,其EZH2Kiapp = 1.2±0.2nM。前列腺癌细胞系LNCaP对EZH2抑制最敏感,GSK343的生长IC50值为2.9μM[1]。 |
| In Vivo | 与对照相比,GSK343(5mg/kg)处理的小鼠显示出显著抑制的肿瘤生长。 GSK343处理组的平均肿瘤体积和重量显著降低。早在植入后20天,相对于对照组,在GSK343处理的群组中观察到肿瘤生长显著减少;这种差异在研究的其余部分持续存在。此外,与对照组相比,异种移植模型中GSK343处理的动物显示E-钙粘蛋白的信使RNA水平显著增加,但波形蛋白信使RNA水平显著降低[2]。 |
| 激酶实验 | 从100%DMSO中的固体制备10mM GSK343原液。以384孔格式(1:3稀释,第6和18列为等体积DMSO对照)制备11点连续稀释母板,并使用声学分配技术分配至测定准备板,以产生100nL GSK343和DMSO对照标记。 。测定添加物由等体积添加的10nM EZH2组成,并且使用多滴组合分配将底物溶液(5μg/ mL HeLa核小体和0.25μM[3H] -SAM)分配到测定板中。将反应板温育1小时,并用等体积添加的含有0.1mM未标记SAM的0.5mg/mL PS-PEI成像珠(RPNQ0098)淬灭。将板密封,暗适应30分钟,并使用Viewlux成像仪获得5分钟终点发光图像。使用ActivityBaseXE分析诸如Z’和背景信号以及剂量响应曲线的平板统计。使用与EZH2相同的384孔SPA测定法评估EZH1的体外生物化学活性作为5成员PRC2复合物的一部分。缓冲组分,试剂分配,GSK343平板制备,淬灭条件和数据分析对于EZH1和EZH2是相同的,最终测定浓度为20nMEZH1,5μg/ mL HeLa核小体和0.25μM[3H] -SAM。进一步的数据分析,pIC50枢轴和可视化由TIBCO Spotfire [1]启用。 |
| SMILES | O=C(C1=CC(C2=CC(N3CCN(C)CC3)=NC=C2)=CC4=C1C=NN4C(C)C)NCC5=C(CCC)C=C(C)NC5=O |
| 靶点 | Histone Methyltransferase |
| 动物实验 | 小鼠[2]使用6周龄雌性裸BALB/c小鼠。为了研究EZH2抑制剂GSK343的作用,在肿瘤体积达到100mm 3后,每隔一天腹膜内注射5mg/kg的100μL磷酸盐缓冲盐水至BALB/c裸鼠(n = 6)。在该分析中,阴性对照组(n = 6)接受盐水。 40天后,杀死小鼠,手术切除皮下肿瘤,称重,拍照,切片,并在10%福尔马林中固定。通过实时逆转录聚合酶链反应测量肿瘤中E-钙粘蛋白,N-钙粘蛋白和波形蛋白的表达水平。 |
| 细胞实验 | 为了解释癌细胞系中不同的倍增率,通过检查它们在384孔板中在6天内以大范围的接种密度进行生长,对所有细胞系凭经验确定最佳细胞接种。然后将细胞以最佳接种密度接种并使其粘附过夜。用20点2倍稀释系列的GSK343或0.147%DMSO(载体对照)一式两份处理细胞,并在37℃,5%CO 2中温育6天。然后用每孔25μLCellTiter-Glo裂解细胞,用TECAN Safire2酶标仪定量化学发光。另外,在添加GSK343时(T0)收获未处理的细胞板以定量细胞的起始数。处理6天后的CTG值表示为T0值的百分比,并相对于GSK343浓度作图。数据与4参数方程拟合以产生浓度响应曲线,并确定抑制50%生长所需的GSK343浓度(gIC50)[1]。 |
| 数据来源文献 | [1]. Sharad K, et al. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med Chem Lett. 2012 Oct 19;3(12):1091-6. [2]. Ding M, et al. The polycomb group protein enhancer of zeste 2 is a novel therapeutic target for cervical cancer. Clin Exp Pharmacol Physiol. 2015 May;42(5):458-64 |
| 规格 | 5mg 10mg |
GSK343 是一种高效,选择性的 EZH2 抑制剂。