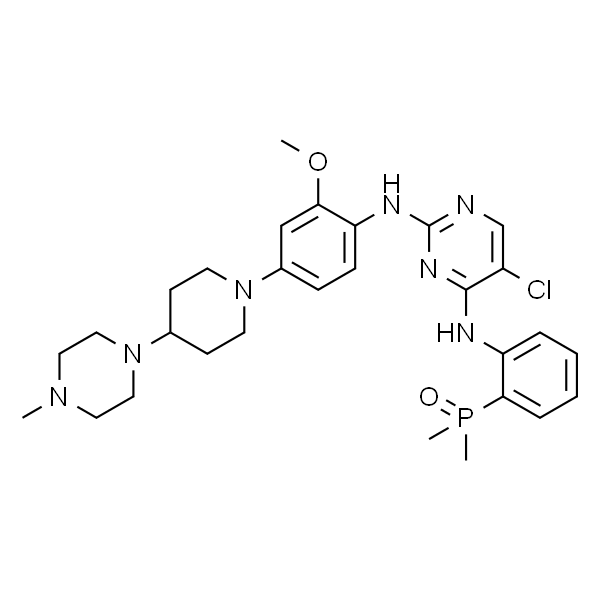
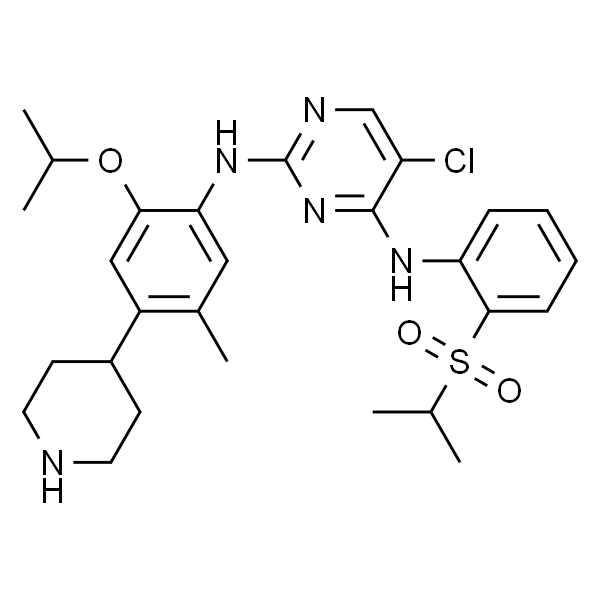
色瑞替尼
货号:
IC2550
品牌:
Jinpan
暂无详情
产品简介
| 有效期 | 2年 |
| 描述 | 是一种有效的ALK抑制剂。 |
| EC | EINECS 811-457-8 |
| MDL | MFCD26142648 |
| 别名 | LDK-378;赛瑞替尼 |
| 英文名称 | Ceritinib |
| CAS | 1032900-25-6 |
| 分子式 | C28H36ClN5O3S |
| 分子量 | 558.14 |
| 储存条件 | 2-8℃ |
| 纯度 | ≥98% |
| 外观(性状) | White to off white Solid |
| 单位 | 瓶 |
| 生物活性 | Ceritinib (LDK378) 是有效且特异性的 ALK 抑制剂,IC50 为 0.2 nM。[1-3] |
| IC50 | 0.2 nM (ALK); 7 nM (InsR); 8 nM (IGF-1R); 23 nM (STK22D); 60 nM (FLT3); 260 nM (FGFR2)[1] |
| In Vitro | Ceritinib(LDK378)也抑制RET(IC50 = 400 nM),FGFR3(IC50 = 430 nM),LCK(IC50 = 560 nM),JAK2(IC50 = 610 nM),极光(IC50 = 660 nM),LYN(50 = 840 nM),EGFR(IC50 = 900 nM)和FGFR4(IC50 = 950 nM)[1]。 Ceritinib(LDK378)保留了对ALK酶活性的高效力,IC50值为200pM,并且仅显示出对46种激酶组中IGF-1R,InsR和STK22D的强抑制,最小选择性为70倍。在用各种激酶转染的Ba / F3细胞中,Ceritinib抑制ALK活性,IC50值为40.7nM,并且对所有其他测试的激酶具有> 100nM的IC 50值。 Ceritinib(LDK378)显示出有效的抗增殖活性,在载有NPM-ALK融合基因的Karpas 299人非霍奇金氏Ki阳性大细胞淋巴瘤中IC50值为22.8 nM,在用NPM-ALK融合转染的Ba / F3细胞中为26 nM基因。 Ceritinib对野生型Ba / F3细胞(IC50>2μM)和用Tel-InsR基因转染的Ba / F3细胞(IC50 = 320 nM)也表现出良好的选择性[2]。 |
| In Vivo | Ceritinib(LDK378)在啮齿动物和非啮齿动物中具有优异的药代动力学特征,口服生物利用度> 50%。 Ceritinib显示剂量依赖性肿瘤生长抑制并且在每天施用的Karpas 299大鼠异种移植模型中实现部分肿瘤消退,但是能够在携带EML4-ALK融合基因的H2228 NSCLC大鼠异种移植模型中实现完全肿瘤消退。在两种模型中,Ceritinib(LDK378)在动物中具有良好的耐受性。 Ceritinib(LDK378)进一步评估其ADME谱,发现在肝微粒体中具有相对良好的代谢稳定性,适度的CYP3A4抑制,在hERG膜片钳实验中一些hERG抑制,IC50值为46μM,但没有QTc证据狗和猴遥测研究中的延长[2]。 |
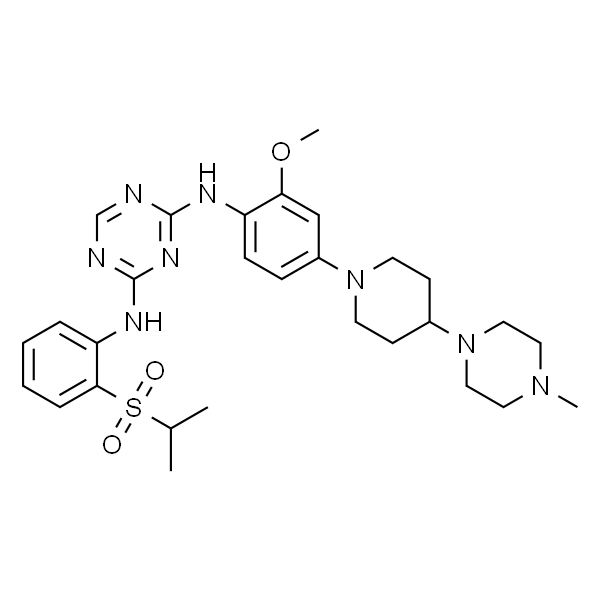
| SMILES | CC(C)OC1=CC(C2CCNCC2)=C(C)C=C1NC3=NC=C(Cl)C(NC4=CC=CC=C4S(=O)(C(C)C)=O)=N3 |
| 靶点 | ALK |
| 动物实验 | 在小鼠,大鼠,狗和食蟹猴中进行体内PK研究。塞立替尼(LDK378)(HCl盐)通过尾静脉以5mg / kg(n = 3)静脉内施用于雄性Balb / c小鼠,并通过管饲法以20mg / kg(n = 3)口服。通过使用相同的制剂,Ceritinib(LDK378)(HCl盐)通过尾静脉以3mg / kg(n = 3)静脉内给予Sprague-Dawley大鼠,并通过管饲法以10mg / kg(n = 3)口服给药。 )。在给药后24小时内按预定时间连续收集血液样品。雄性比格犬分别接受单次静脉内(n = 2)或口服(n = 3)剂量的Ceritinib(磷酸盐)作为5mg / kg的静脉内溶液和20mg / kg的口服悬浮液。雄性食蟹猴接受单次静脉内(n = 2)或口服(n = 3)剂量的Ceritinib(游离碱)作为5mg / kg的静脉内溶液和60mg / kg的口服悬浮液。在给药后144小时,在预定时间收集血浆的血液样品[1]。 |
| 数据来源文献 | [1]. Marsilje TH, et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl [2]. Chen J, et al. LDK378: a promising anaplastic lymphoma kinase (ALK) inhibitor. J Med Chem. 2013 Jul 25;56(14):5673-4. [3]. Tucker ER, et al. Immunoassays for the quantification of ALK and phosphorylated ALK support the evaluation of on-target ALK inhibitors in neuroblastoma. Mol Oncol. 2017 Aug;11(8):996-1006. |
| 规格 | 5mg 10mg |