NG25
货号:
IN0690
品牌:
Jinpan
暂无详情
产品简介
| 含量 | Purity≥98% |
| 别名 | NG-25 |
| CAS | 1315355-93-1 |
| 分子式 | C29H30F3N5O2 |
| 分子量 | 537.58 |
| 纯度 | ≥98% |
| 单位 | 支 |
| 生物活性 | NG25 是一种 TAK1 和 MAP4K2 的双抑制剂,IC50 值分别为 149 nM 和 21.7 nM[1-3]。 |
| IC50 | TAK1:149 nM ; MAP4K2:21.7 nM [1-3] |
| In Vitro | NG25是一种有效的双重TAK1和MAP4K2抑制剂,IC50分别为149 nM和21.7 nM。 NG25还有效抑制几种激酶,如LYN,CSK,FER,p38α,ABL,ARG和SRC,IC50分别为12.9,56.4,82.3,102,75.2和113 nM [1]。 NG25是CpG B-或CpG A刺激的IFNα分泌和CL097刺激的IFNβ分泌的非常有效的抑制剂,完全抑制400nM [2]。 NG25处理以剂量依赖性方式降低所有测试的乳腺癌细胞系的细胞活力。 NG25(2μM)增强Dox对乳腺癌细胞的细胞毒作用[3]。 |
| 激酶实验 | IRF7在大肠杆菌中表达为谷胱甘肽S-转移酶(GST)融合蛋白,在GST和IRF7之间具有PreScission蛋白酶切割位点。通过用PreScission蛋白酶消化,在GST和谷胱甘肽 – 琼脂糖凝胶释放的谷胱甘肽 – 琼脂糖凝胶和IRF7上捕获GST-IRF7。 His6标记的IKKβ和TBK1在昆虫Sf21细胞中以其活性磷酸化形式表达,并通过在次氮基三乙酸镍 – 琼脂糖上的亲和层析纯化。活性GST-IKKα购自Millipore并进行测定。 |
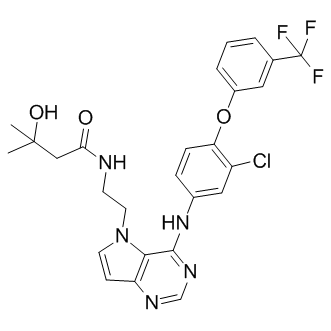
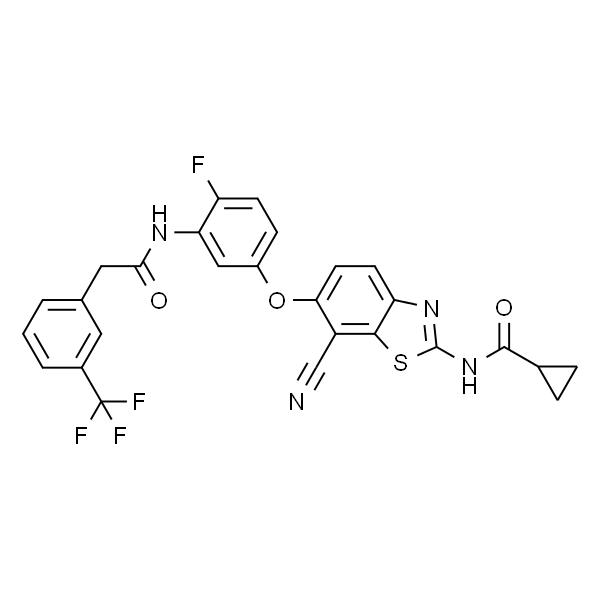
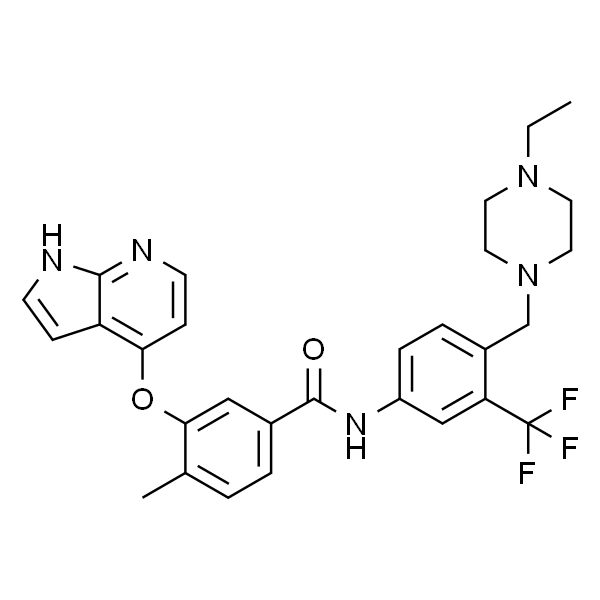
| SMILES | O=C(NC1=CC(C(F)(F)F)=C(CN(CC2)CCN2CC)C=C1)C3=CC(OC4=C5C(NC=C5)=NC=C4)=C(C)C=C3 |
| 靶点 | MAP4K |
| 细胞实验 | 将3.5×105 Gen2.2细胞或Flt3-DC在96孔板中孵育1小时,不含或不含指定浓度的抑制剂,然后用1μMCpG(A型或B型)或1μg/ mL CL097刺激或R848。 5或12小时后,收集细胞培养物上清液,通过离心澄清,并在-80℃冷冻直至分析细胞因子水平。对于细胞活力测定,未受刺激的细胞在不存在或存在抑制剂的情况下孵育12小时。然后固定细胞并通过流式细胞术分析活细胞的百分比。 |
| 数据来源文献 | [1]. Tan L, et al. Discovery of type II inhibitors of TGFβ-activated kinase 1 (TAK1) and mitogen-activated protein kinase kinase kinase kinase 2 (MAP4K2). J Med Chem. 2015 Jan 8;58(1):183-96. [2]. Pauls E, et al. Essential role for IKKβ in production of type 1 interferons by plasmacytoid dendritic cells. J Biol Chem. 2012 Jun 1;287(23):19216-28. [3]. Wang Z, et al. TAK1 inhibitor NG25 enhances doxorubicin-mediated apoptosis in breast cancer cells. Sci Rep. 2016 Sep 7;6:32737 |
| 规格 | 2mg 5mg 10mg |
NG25 是一种 TAK1 和 MAP4K2 的双抑制剂。